全国热线:400 0087 916
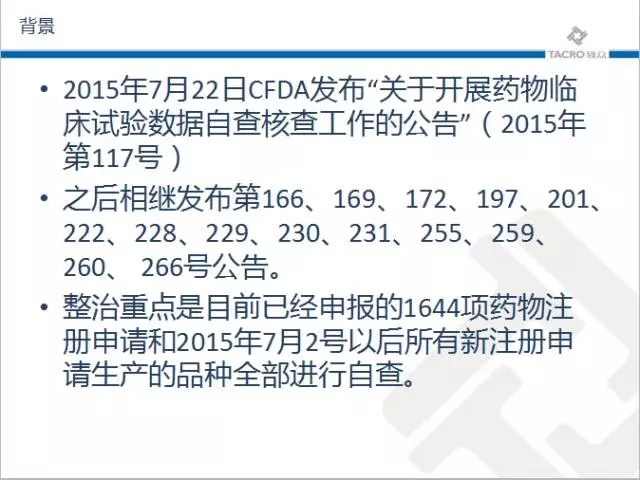
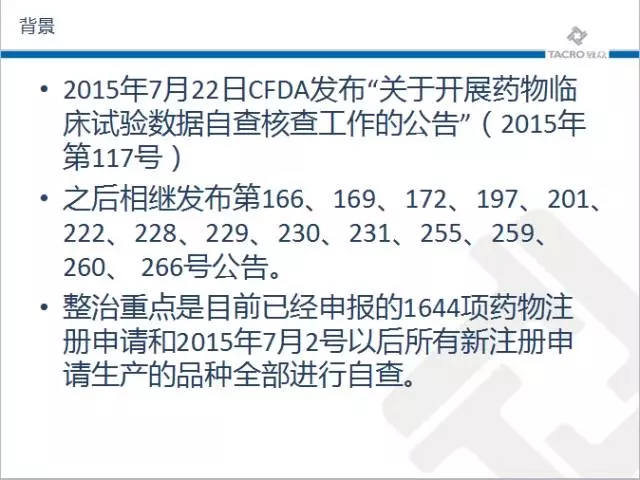
随着针对药品的临床试验进展的如火如荼,2015年7月22日CFDA发布“关于开展药物临床试验数据自查核查工作的公告”(2015年第117号)之后相继发布第166、169、172、197、201、222、228、229、230、231、255、259、260、 266号公告。整治重点是目前已经申报的1644项药物注册申请和2015年7月2号以后所有新注册申请生产的品种全部进行自查。
CFDA将要采取的措施中,第一点就是:整肃临床试验数据造假“潜规则”,净化研发生态环境,针对医疗器械临床核查的呼声也日益高涨。2016年2月27日,CFDA公布了一起医疗器械临床数据造假的案件, 官网发布《总局关于对日本富士瑞必欧株式会社的乙型肝炎病毒核心相关抗原(HBcrAg)检测试剂盒不予注册的公告(2016年第44号)》。为了响应国家局的号召,2016年3月10日,北京市CFDA发布《北京市食品药品监督管理局关于开展医疗器械临床试验自查的通告》。
2016年中国医疗器械临床数据核查工作的大幕已经开启,企业在此轮核查风暴下究竟该如何从容应对?为帮助园区医疗器械企业按照新版医疗器械临床试验质量管理规范进行有效、有序的数据自查以降低核查风险成本,武汉光谷生物城生物医药产业园、武汉光谷加速器投资发展有限公司联合武汉致众科技股份有限公司于昨日在武汉光谷生物城生物医药园共同举办“总局核查风暴下 医疗器械临床试验数据核查要点解析及应对策略交流会”。
上一篇:如何做一个规范的医疗器械经营企业?(附:武汉市医疗器械经营许可证办理指南)
下一篇:【原创】电磁兼容常用器件的选用
活动报名