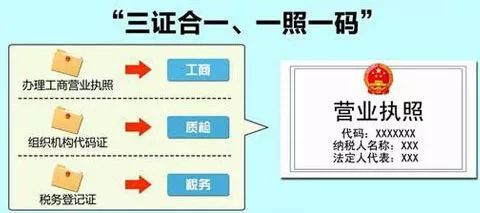
工商注册
现如今,公司实行注册资本认缴制和三证合一登记制度,注册流程大为简化,注册费用大为节约,注册周期也大为缩减。
但在公司注册前,需认真思考经营范围,前置考虑未来产品生产范围,避免后续再做变更。
产品开发设计
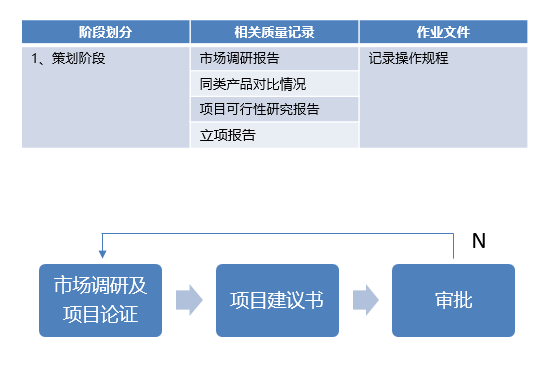
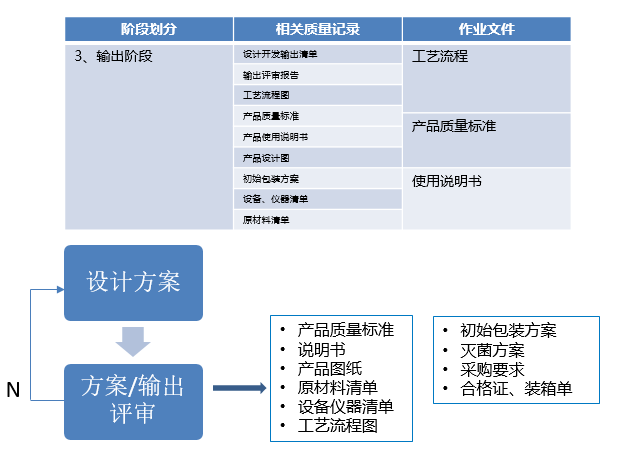
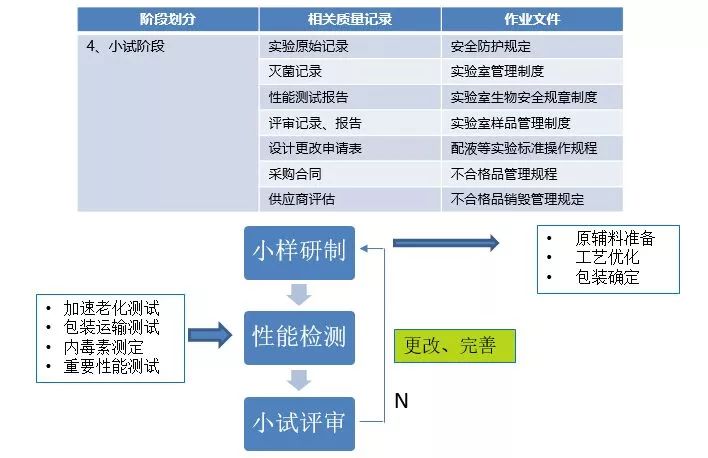
产品设计开发可分7个阶段:策划阶段、设计输入阶段、设计输出阶段、小试阶段、中试阶段、定型阶段、注册资料准备阶段。
策划阶段
输出阶段
小试阶段
中试阶段
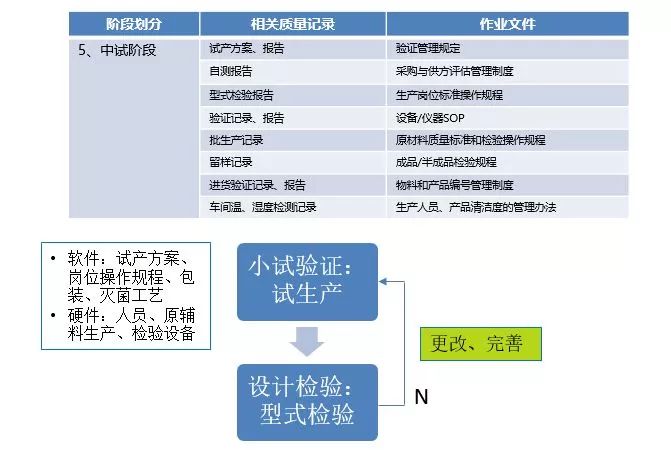
定型阶段
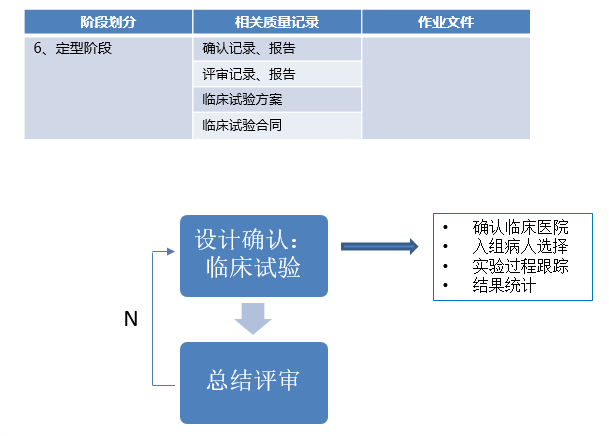
注册资料准备阶段与产品注册阶段
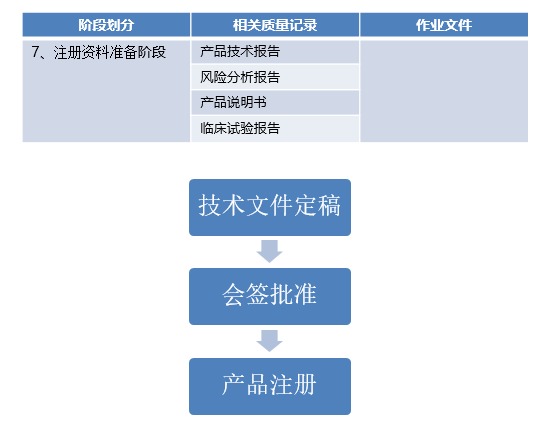
创新医疗器械申报
体系建立法规背景
《关于发布医疗器械生产质量管理规范附录无菌医疗器械的公告》(2015年第101号)、《关于发布医疗器械生产质量管理规范附录植入性医疗器械的公告(2015年第102号)及《关于发布医疗器械生产质量管理规范附录体外诊断试剂的公告》(2015年第103号),于2015年10月1日起正式施行。
《关于第一类、第二类医疗器械生产企业实施医疗器械生产质量管理规范有关工作的通知》(食药监办械监〔2017〕120号)与2017年9月1日发布。
医疗器械生产企业需严格按照法规要求建立质量管理体系。而对于初创新公司除了体系建立,还需要面临厂房选址和设计、建设等问题。
体系建立厂房规划
对于厂房问题,需要根据产品管理类别进行考量。
非无菌产品要求相对较低。若为无菌、体外诊断类产品,则应严格按照法规和标准选址,远离有污染的空气和水(如远离铁离、码头、机场、交通要道以及散发大量粉尘、屠宰场、染料等);
对厂房的设计和装修,必须请专业的团队和公司来设计和施工,如行政区、生活区和辅助区不得相互防碍影响,空气洁净级别不同的洁净室(区)之间的静压差应大于5帕,洁净室(区)与室外大气的静压差应大于10帕,空气洁净度级别进行合理布局,人流、物流走向应当合理,避免交叉污染,注意洁净室的水池或地漏等;
虽然委托专业公司负责,但整个过程,都需要专业体系人员进一步把关,避免整改,比如消防、环评等通不过等。
人员配置
必备岗位人员:生产负责人1名、研发部负责人1名、质量负责人1名、专职检验员2名、总经理1名;可兼任岗位人员:管理者代表1名,可由质量负责人兼任;采购部负责人1名,可由研发老大兼任;销售部负责人1名,可由产品经理或总经理兼任;行政部负责人1名,也可让总经理兼任;内审员2名,持有内审资格证的即可兼任,但2人不得在同部门;生产人员1名以上;
如此算来十多个岗位,兼任下来,至少应有6人以上
体系认知
首先,质量管理体系是个系统工作,要有系统的观念和思维。文件及记录仅仅是整个系统的一部分,一个子系统而已,此外还有关键的管理控制子系统,设计控制子系统,生产制造子系统。需要与公司的培训系统,绩效系统,营销系统等等结合互动。
当前法规的目标是确保质量管理体系的有效性以持续生产安全有效的医疗器械产品。基于此,法规监管并不希望企业三天两头修改技术文件,工艺文件等,而是应保障在当前法规要求下,可持续生产安全有效医疗器械产品。若日常工作中就对法规要求有所思虑,那么质量管理体系法规符合性相对是较好的。
再者,实施质量管理体系的终极目的,不是为了一张认证证书,而是降低风险。对企业来说,若证书拿到就万事大吉,那么企业发展必定不会长远。老老实实的做好基础,做好系统管理。更容易达到实施体系的目的:预防为主,降低风险。
注册检验法规背景
《医疗器械分类界定管理办法》明确规定,申请第二类、第三类医疗器械注册,应当进行注册检验。医疗器械检验机构应当依据产品技术要求对相关产品进行注册检验。注册检验样品的生产应当符合医疗器械质量管理体系的相关要求,注册检验合格的方可进行临床试验或者申请注册。办理第一类医疗器械备案的,备案人可以提交产品自检报告。
注册检验内容
医疗器械注册检验时检验机构(必须是CFDA认可有资质的机构)会依据企业所提供给的医疗器械产品技术要求做相应的检验,检验内容主要包括安规性能检验,EMC电磁兼容性检验(有源产品需要,无源产品不需要),生物相容性检验等。
检测周期
自2017年4月1日起,包括医疗器械产品检验费在内的多项事业性收费取消征收,这里包括医疗器械注册检验收费。
据统计,CFDA认可的全国医疗器械检验机构共有53家,各医疗检验机构对这一政策事先并无充分预案,但又必须按时执行。政策貌似助力行业发展,但短期内却给行业发展带来诸多混乱。企业送检就要排长队,过去60个工作日即可完成的,现在200个工作日也未必能完成。有些机构只接受本省样品,跨省不接受,因为取消收费以后,经费均由本省承担,没有充足的资源对外提供服务。
企业能做的就是在产品开发立项过程中引入专业法规人员把关,协助研发工程师在设计之处就能明确遵循的标准和法规要求,降低后期整改难度。并在送检前的开发阶段做好充分的验证测试,顺利提高检测通过率。
发展预测
不难发现,中国的医疗器械注册检验制度体系正在重构,并逐渐形成如下趋势:
首先,制造商应当注重构建自己的自检体系。因为放松管制意味着制造商将承担更多的主体责任;
其次,官方的检验机构正在重新定位,明确自己新的职责,如继续承担监督抽验的任务,承担强制性标准实施任务等等;
最后,第三方检验机构迎来新机遇。第三方检验机构一直想在注册检验领域为企业提供服务。过去没有机会,现在机会出现了,大量第三方检测机构未来会有很多的机会参与到整个检验体系的构建当中,分得一杯羹。检验资源增加了,企业会有更多的自主选择权,这是好事。未来检验体系的改革一定是有利于产业发展的。
临床评价法规背景
《医疗器械注册管理办法》(国家食品药品监督管理总局令第4号)
《医疗器械临床评价技术指导原则》(2015年第14号通告)
《医疗器械临床试验质量管理规范》(国家食品药品监督管理总局/中华人民共和国国家卫生和计划生育委员会令第25号)
《关于医疗器械临床试验备案有关事宜的公告》(2015年第87号)
《免于进行临床试验的第二类医疗器械目录》(2014年第12号通告)——488个II类产品
《免于进行临床试验的第三类医疗器械目录》(2014年第13号通告)——79个III类产品
《第二批免于进行临床试验医疗器械目录》(2016年第133号通告)——267个II类(含15个IVD),92个III类产品
《需进行临床试验审批的第三类医疗器械目录》(2014年第14号的通告)——8个III类产品
《第三批免于进行临床试验医疗器械目录》(2017年170号)——116个II类IVD,11个III类产品,37个II类产品
临床评价路径三种途径:
对列入《免于进行临床试验的医疗器械目录》中的产品,有条件的免于临床试验;
对于同品种医疗器械临床试验或临床使用获得的数据进行分析评价;
按照《医疗器械临床试验质量管理规范》开展临床试验。
实施建议
产品若不在目录内,则只能通过临床试验或临床评价两个途径。对于新公司首款产品,条件允许的话,建议做临床试验。
首先,免临床途径很难拿到经验数据和对比资料的授权;
其次,首款产品注册上市后,其他产品注册也会很快启动,便于后续产品临床工作开展。
关于临床试验工作,对于初创团队而言,建议委托第三方有实力、专业的CRO团队,利于更快、更好的推进临床进度。
产品注册申报
撰写准备产品综述资料、研究资料、生产制造信息、临床评价资料、产品风险分析资料、产品技术要求、产品注册检验报告、说明书和标签样稿等资料清单,整理递交CFDA。
生产许可申请法规背景
医疗器械先注册后许可:新办企业会面临一个特殊时期,拿到注册证不能马上销售,需要申请“生产许可证”。
《医疗器械监督管理办法》明确规定:从事第二类、第三类医疗器械生产的,生产企业应当向所在地省、自治区、直辖市人民政府食品药品监督管理部门申请生产许可并提交其符合本条例规定条件的证明资料以及所生产医疗器械的注册证。
受理生产许可申请的食品药品监督管理部门应当自受理之日起30个工作日内对申请资料进行审核,按照国务院食品药品监督管理部门制定的医疗器械生产质量管理规范的要求进行核查。
对符合规定条件的,准予许可并发给医疗器械生产许可证;对不符合规定条件的,不予许可并书面说明理由。
申请周期
生产许可申请,法规规定为30个工作日,即1个月的时间。
若现场审核无重大缺陷,并整改顺利,基本上2-3个月即可拿到生产许可证。在此阶段,公司可提前预热,做好市场推广,参展试用,但切记不可销售。
在申请生产许可期间,公司基本上全部流程、文件都已形成,人员也已全部到位。
生产许可证与注册证差异(1)医疗器械产品注册证
•医疗器械产品注册证是医疗器械产品上市销售的合格证明。•第一类医疗器械产品备案,向市级食品药品监督管理局提交备案资料。第二类医疗器械产品注册,向所在地省、自治区、直辖市食品药品监督管理部门提交注册申请资料。•第三类医疗器械产品注册,注册申请人应当向国家食品药品监督管理局(CFDA)提交注册
申请资料-医疗器械生产许可证
•医疗器械生产许可证是医疗器械生产企业获得医疗器械产品生产的资质证明。•从事第一类医疗器械生产的,向市食品药品监督管理局备案。•从事第二类、第三类医疗器械生产的,省、自治区、直辖市食品药品监督管理局申请生产许可并提交相应的证明资料以及所生产医疗器械的注册证。
© 2018 - 2020, Wuhan Tacro Technology Co.,Ltd All Rights Reserved.

