上次我们讲了伦理审查的内容(器械伦理审查要点有哪些?http://mp.weixin.qq.com/s?__biz=MzA3OTA4NzQzMA==&mid=401842399&idx=1&sn=ee9d704d4edb789555ec2c79b6ab6074#rd),这不,今天北京市药监局发布了全国第一个临床自查的通告:《北京市食品药品监督管理局关于开展医疗器械临床试验自查的通告》(上篇微信),自查内容第一、二条便是“1.临床试验机构条件及合规性。2.受试者知情同意、伦理审查情况。” 今天我们来说一下医疗机构伦理审查的步骤和一般情况下伦理委员会批准的标准。
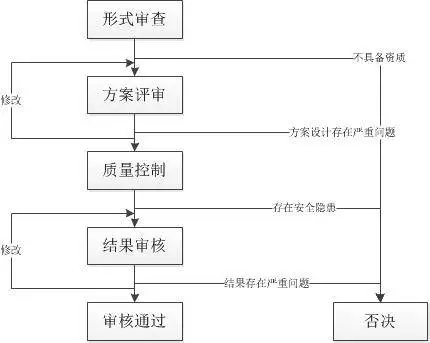
一般而言,医疗机构中医疗器械临床试验的伦理审查主要分几个步骤:形式审查;方案评审;质量控制;结果审核。
01
形式审查
主要审查的内容包括:试验产品是否满足国家食品药品监督管理局要求的开展临床试验的前提条件,比如:产品具有产品技术要求和自测报告;产品具有国家食品药品监督管理总局指定的检测机构出具的型式检测报告,且结论为合格等;
另外,研究机构和研究者是否具有承担该项试验的资格和条件,是否具有合适的试验负责人和研究团队。
形式审查是对开展临床试验研究的各方资质和条件进行审查,不符合要求的是不能开展临床试验的。
02
方案评审
方案评审是伦理委员会成员对医疗器械临床试验方案进行评审。医疗器械临床试验开展前必须制定临床试验方案,医疗器械临床试验方案应当最大限度地保障受试者权益、安全和健康,所以伦理委员会成员需要对方案各方面包括入排标准、评价标准、不良反应的观察和处理等进行审核并提出意见和建议。一旦通过伦理委员会审查则临床试验必须按照该试验方案进行。若试验进行过程中发现问题需要修改,必须经伦理委员会再次审查并批准。如果试验方案未通过伦理审查,临床试验将不能开展。
03
质量控制
质量控制是对临床试验开展过程中试验质量的监督和管理。通过对病例入组、试验操作、数据测量和记录等进行定期检查和不定期抽查,及时发现和纠正临床试验过程中的问题,以确保临床试验的质量。对于存在重大违规和严重安全隐患的临床试验,伦理委员会有权利责令研究者停止或终止临床试验。
04
结果审核
临床试验报告应在医疗器械临床试验完成后按医疗器械临床试验法规的要求和规定的格式撰写,并且需要和临床试验方案保持一致。伦理委员会对试验结果和试验报告进行审核,主要包括:试验数据是否真实、可信;试验数据分析是否合理、详实;结论推断是否合适、恰当;试验过程中是否发生不良事件,如何处理及解释;受试者利益是否得到保障。若经伦理委员会审核后决定试验报告需要修改,则修改后的试验报告必须经伦理委员会再次审核直至通过。
医疗器械临床试验的伦理批准标准
1)试验产品满足开展临床试验的前提条件;
2)试验实施者或研究者具备开展该研究的资质:
3)临床试验设计科学、合理,试验方案规范、细致,试验具有可行性;
4)受试者在试验过程中的安全风险最小化;
5)受试者的安全风险相对于对受试者的预期受益或预期社会效益的重要性来说是合理的;
6)受试者的选择是在公平、自愿、充分知情条件下进行的;
7)每位受试者或其法定代理人对参加的临床试验知情同意,且知情同意具有相应的文件证明,
8)有充分的安全监查计划保证受试者的安全;
9)有充分的规定保护受试者的隐私;
10)必要时,有保护弱势群体受试者的措施。
© 2018 - 2020, Wuhan Tacro Technology Co.,Ltd All Rights Reserved.

