医疗器械产品注册
技术审评报告
(境内)
产品中文名称:血管重建装置
产品管理类别:第三类
申请人名称: 微创神通医疗科技(上海)有限公司
国家食品药品监督管理总局
医疗器械技术审评中心
基本信息
一、申请人名称
微创神通医疗科技(上海)有限公司
二、申请人住所
上海市浦东新区广丹路222 弄16 幢
三、生产地址
上海市浦东新区广丹路222 弄16 幢
产品审评摘要
一、产品概述
(一)产品结构及组成
该产品由支架系统和微导管系统组成。支架系统由支架(见图1)和输送器组成,输送器包括输送导丝和导入鞘;微导管系统由微导管和塑形针组成。支架由镍钛合金丝编织而成,且每个支架均由两根铂/铱显影丝贯穿整个支架,采用自扩张方式释放。输送导丝为具有显影标记的介入导丝,其芯丝材料为镍钛合金,远端缠绕一段304V 不锈钢组成的柔软绕丝,输送导丝外涂有亲水涂层。输送导丝连同支架一起,预装在导入鞘内,通过输送导丝上的输送膜将支架推入到微导管中,并最终从微导管中释放。微导管具有半刚性的近端杆和柔软的远端,头端具有不透X 射线标记。
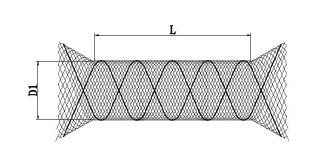
图1 支架示意图
(二)产品适用范围
该产品适用于颈内动脉及椎动脉未破裂囊性动脉瘤的患者,动脉瘤瘤颈≥4mm 且瘤体最大径≥10mm,靶病变血管直径2.0mm-6.5mm。
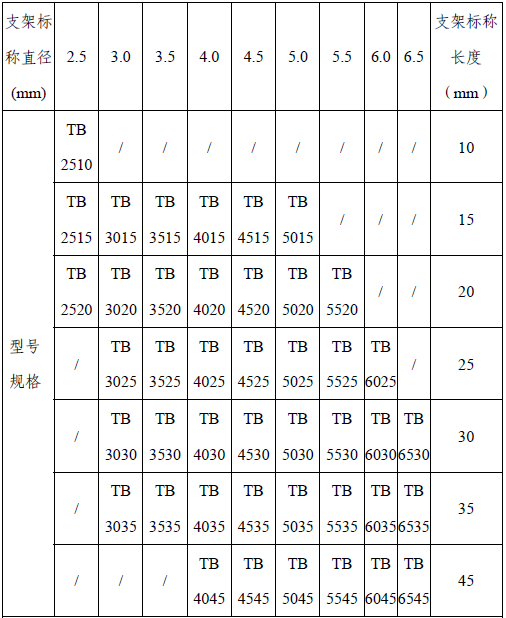
(三)型号/规格
表1 型号规格表
(四)工作原理
在医学影像设备的监护下,通过输送导丝的推送,将支架在微导管内输送至靶病变部位,采用自扩张方式释放支架,通过支架的密网孔结构改变动脉瘤的血流动力学,减少流入动脉瘤囊的血流,诱发动脉瘤内血栓形成,促进瘤颈部的内膜增生,达到治疗颅内动脉瘤目的。
二、临床前研究摘要
(一)产品性能研究
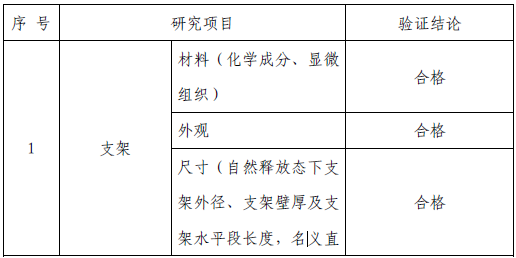
1. 产品技术要求研究
技术要求研究项目如表2 所示:
表2 技术要求研究摘要
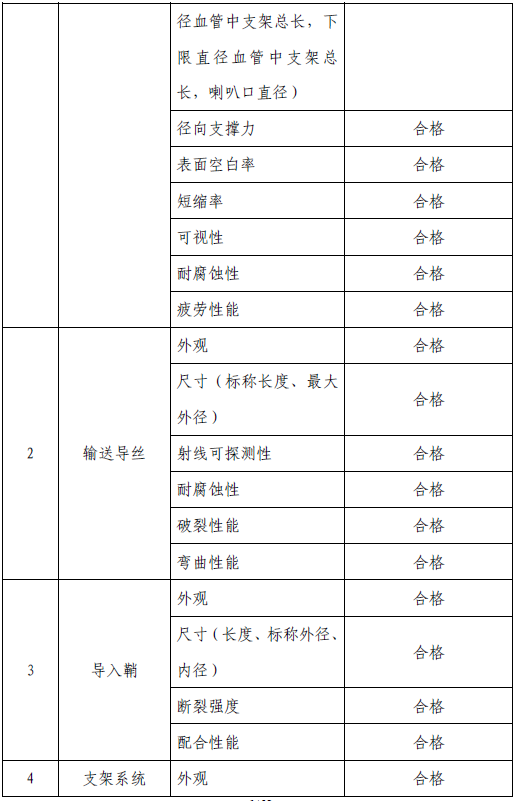
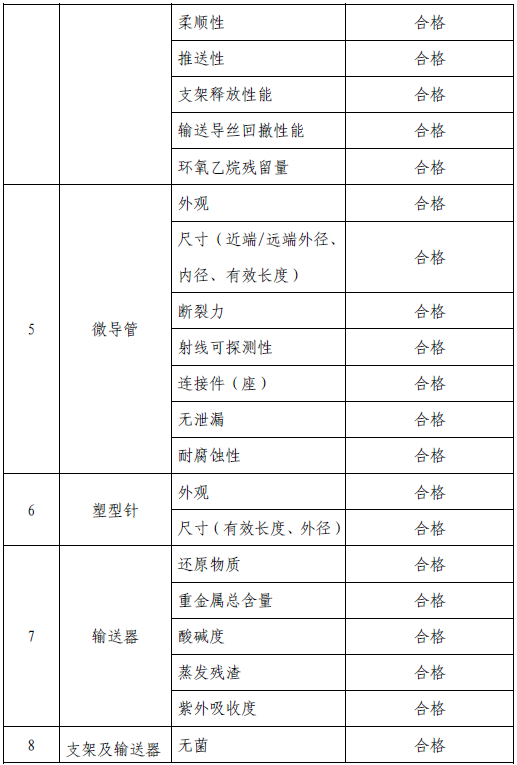
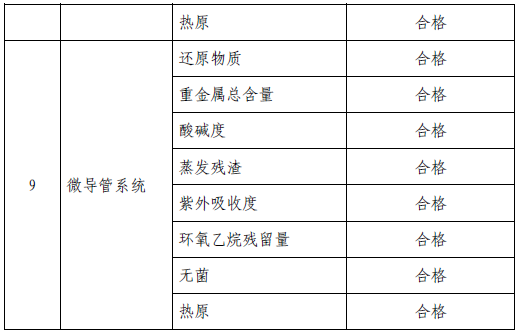
2. 产品性能评价
产品性能评价包括模拟使用、MRI 兼容性、支架有限元分析、镍离子析出安全性、血流动力学等研究,结果表明产品符合设计输入要求。
(二)生物相容性
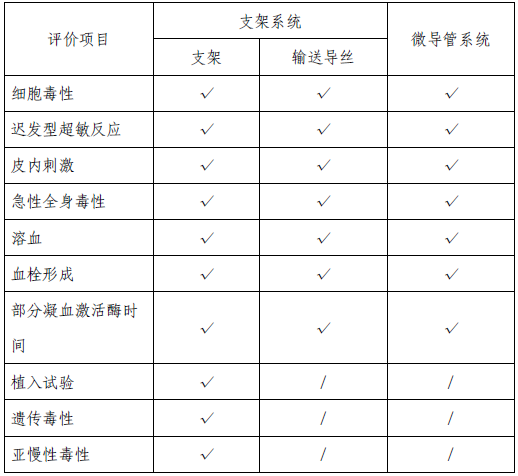
该产品包含支架系统和微导管系统两部分,支架系统中支架为植入器械,与循环血液长期接触;支架系统中输送器(含输送导丝和导入鞘)为外部接入器械,与循环血液短期接触;微导管系统为外部接入器械,与循环血液短期接触。申请人依据GB/T 16886 系列标准对植入器械及外部接入器械分别进行了生物相容性评价,产品生物相容性风险可接受,具体评价项目
详见表3。
表3 生物相容性评价内容表
(三)灭菌
该产品采用环氧乙烷灭菌,无菌状态提供。申请人提供了灭菌过程确认报告,证明无菌保证水平为10-6。环氧乙烷残留量不大于10μg/g,2-氯乙醇残留水平不超过9mg/件。
(四)产品有效期和包装
该产品货架有效期两年。申请人提供了货架有效期验证报告,验证实验为实时老化验证,包括产品稳定性、包装完整性验证。
(五)动物研究
该实验目的为评价血管重建装置在成功制作动脉瘤的新西兰大白兔动物模型中产品使用性能及安全有效性。每只动物植入两枚支架,植入即刻、植入后4 周、植入后3 个月分别进行
观察,评估动脉瘤治疗效果和对穿支血管影响等。评价指标包括:
器械可操作性:评价输送系统到达靶病变位置的能力、操作性、可视性、回撤能力、释放支架的准确性和有效性、支架的可视性、尺寸的适合性、位置、完整性和功能性以及支架在弯曲血管的贴壁性能、输送系统和导入鞘的止血性。器械生物安全性:血管内血栓形成、炎症反应、支架内皮化情况以及支架植入位置附近是否有局部出血、组织坏死等。器械有效性:观察动脉瘤闭塞率和支架所覆盖分支通畅率。
动物实验结果表明,产品达到预期设计要求。
三、临床评价摘要
该产品以临床试验方式进行临床评价。临床试验目的为评价申报产品用于治疗“靶病变血管直径2.0mm~6.5mm,瘤颈≥4mm 且瘤体最大径≥10mm 的颈内动脉及椎动脉未破裂囊性动脉瘤”的安全有效性。临床试验为前瞻性、多中心、随机对照的优效性设计,对照组采用血管重建装置合并弹簧圈。
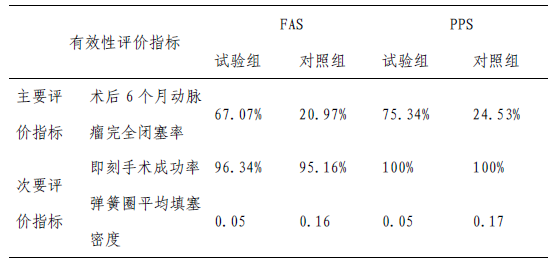
该临床试验在12 家临床机构开展,实际入组144 例(试验组82 例/对照组62 例)患者。主要研究终点为术后6 个月动脉瘤完全闭塞率,次要有效性指标为即刻手术成功率,术后瘤体
内弹簧圈填塞密度。安全性评价指标为术后30 天、术后90 天、术后1 年受试者死亡或病变血管相关性卒中,目标动脉瘤出血、不良事件发生率,术后6 个月支架内狭窄和支架内血栓发生率。
全分析集(FAS)包含患者144 例,其中试验组82 例,对照组62 例;符合方案集(PPS)包含患者126 例,其中试验组73 例,对照组53 例;安全集(SS)包含患者144 例,其中试验组82 例,对照组62 例。主要/次要有效性指标结果如表4 所示。
结果表明,试验组和对照组术后6 个月动脉瘤完全闭塞率有显著差异,95%可信区间估计表明优效性研究假设成立。
表4 临床试验主要/次要有效性评价指标结果
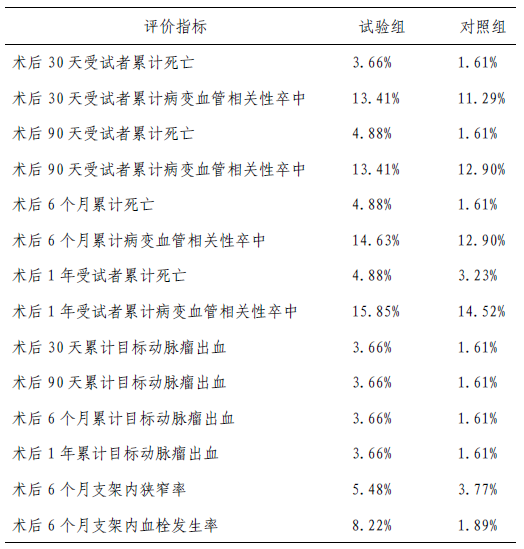
安全性指标分析如表5 所示,死亡、病变血管相关性卒中、目标动脉瘤出血、支架内狭窄、支架内血栓发生率组间无统计学差异。
表5 临床试验安全性评价指标结果(SS 集)
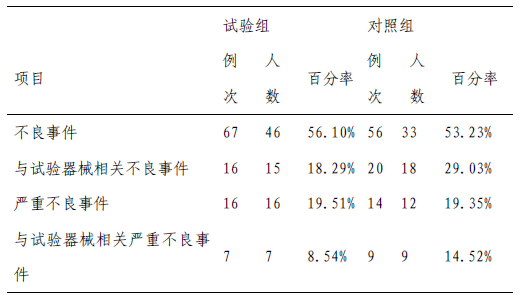
不良事件发生情况如表6 所示,试验组与对照组间无统计学差异,与器械相关性分析结果为试验组与对照组间无统计学差异。
表6 不良事件总体情况
该产品在临床应用中通过改变患者动脉瘤腔内的血流动力学实现动脉瘤的闭塞,可能伴随急性血栓、血管痉挛、早期或迟发性动脉瘤破裂、载瘤动脉闭塞/狭窄、分支动脉闭塞/血流
缓慢、延迟愈合或长期不愈、出血等风险。根据申请人提供的申报资料,经综合评价,在目前认知水平上,认为该产品为患者带来的受益大于风险。
四、风险分析及说明书提示
参照《YY/T 0316-2016 医疗器械风险管理对医疗器械的应用》,对该产品进行风险分析。对目前已知及可预测风险采取了风险控制措施,经综合评价,在目前认知水平上,认为该产品上市带来的获益/受益大于风险。为保证用械安全,需在说明书中提示以下信息:
(一)明确的产品适用范围
(二)警示及注意事项
1.本产品已环氧乙烷灭菌处理;
2.本产品为一次性使用产品,请勿重复灭菌或重复使用;
3.使用前如发现包装已打开、破裂、泄露或已超过灭菌有效期,请勿使用;
4.应由接受过必需的介入训练(尤其是颅内支架植入手术培训)的有资质专业人士使用本产品完成手术的操作;
5.对镍钛(镍钛锘)过敏的患者,植入本品可能会发生过敏反应;
6.不要让系统接触有机溶剂(如酒精等);
7.在使用之前,仔细检查无菌包装以及系统组件,以确保它们在运输中没有被破坏。不要使用扭结或者已破坏的组件;
8.请与配套的微导管配合使用;
9.塑型针不能保留在导管内;只能用蒸汽进行塑型;如果头端有损坏,切勿使用;
10.当输送血管重建装置遇到阻力时,不要试图推进或回撤,直到在可视条件下找出产生阻力的原因;
11.当微导管进入人体后,微导管的移动一定要在射线监测下进行;
12.如果发现微导管堵塞,应当更换一个新的导管或取出导管检查堵塞原因;
13.在动脉瘤处放置多个支架会增加发生缺血性并发症的危险;
14.选择合适的支架尺寸,使其释放后的直径尽可能接近于载瘤动脉的直径,如果选择错误的支架尺寸,可能导致支架定位不准确、释放不完全或者支架移位;
15.支架在释放过程中会大幅短缩,所以在支架释放时应考虑支架短缩的发生;
16.如果输送导丝在微导管中不能移动,应该同时将输送导丝和微导管作为整体移动;
17.做各种动作时(从包装里取出支架及通过导管),小心避免接触支架,因为这会造成支架移位或损坏;
18.血管角度过大时应慎重使用;
19.硬膜内动脉瘤需同时结合弹簧圈使用。
(三)禁忌症
1.对造影介质过敏的患者;
2.患者有明确镍钛合金材料过敏史;
3.被活跃的细菌感染的患者;
4.抗血小板或/和抗凝治疗禁忌的患者;
5.破裂动脉瘤;
6.不适宜支架输送和展开的病变(如病变血管过于迂曲、支架无法达到病变部位、复发动脉瘤的靶血管中存在狭窄或未完全扩张的支架等)。
综合评价意见
该申报产品属按照《创新医疗器械特别审批程序(试行)》审批项目,编号2015123。申请人的注册申报资料符合现行要求。
依据《医疗器械监督管理条例》(国务院令第680 号)、《医疗器械注册管理办法》(国家食品药品监督管理总局令2014 年第4号)等相关医疗器械法规与配套规章,经系统评价后,建议准予注册,同时产品上市后应进一步观察真实世界中远期安全性和有效性。
© 2018 - 2020, Wuhan Tacro Technology Co.,Ltd All Rights Reserved.

